
Each dispersible tablet contains Deferasirox 250 mg and 500 mg.
V03AC03 — Deferasirox
Deferasirox is an iron chelator and the first oral medication FDA approved for chronic iron overload in patients receiving long term blood transfusions.
For the treatment of chronic iron overload due to blood transfusions (transfusional hemosiderosis) in patients 2 years of age and older.
Deferasirox is an orally active chelator that is selective for iron (as Fe3+). It is a tridentate ligand that binds iron with high affinity in a 2:1 ratio. Although deferasirox has very low affinity for zinc and copper there are variable decreases in the serum concentration of these trace metals after the administration of deferasirox. The clinical significance of these decreases is uncertain.
Two molecules of deferasirox are capable of binding to 1 atom of iron. Deferasirox works in treating iron toxicity by binding trivalent (ferric) iron (for which it has a strong affinity), forming a stable complex which is eliminated via the kidneys.
Each 10 ml vial contains gemcitabine (as gemcitabine hydrochloride) 200 mg and 1000 mg.
L01BC05 — Gemcitabine
Gemcitabine is a nucleoside analog used as chemotherapy. As with fluorouracil and other analogues of pyrimidines, the drug replaces one of the building blocks of nucleic acids, in this case cytidine, during DNA replication. The process arrests tumor growth, as new nucleosides cannot be attached to the “faulty” nucleoside, resulting in apoptosis (cellular “suicide”).
Gemcitabine is used in various carcinomas: non-small cell lung cancer, pancreatic cancer, bladder cancer and breast cancer. It is being investigated for use in oesophageal cancer, and is used experimentally in lymphomas and various other tumor types.
Gemcitabine is an antineoplastic anti-metabolite. Anti-metabolites masquerade as purine or pyrimidine – which become the building blocks of DNA. They prevent these substances becoming incorporated in to DNA during the “S” phase (or DNA synthesis phase of the cell cycle), stopping normal development and division. Gemcitabine blocks an enzyme which converts the cytosine nucleotide into the deoxy derivative. In addition, DNA synthesis is further inhibited because Gemcitabine blocks the incorporation of the thymidine nucleotide into the DNA strand. It demonstrates dose-dependent synergistic activity with cisplatin in vitro. In vivo, gemcitabine showed activity in combination with cisplatin against the LX-1 and CALU-6 human lung xenografts, but minimal activity was seen with the NCI-H460 or NCI-H520 xenografts. Gemcitabine was synergistic with cisplatin in the Lewis lung murine xenograft. Sequential exposure to gemcitabine 4 hours before cisplatin produced the greatest interaction.
Gemcitabine inhibits thymidylate synthetase, leading to inhibition of DNA synthesis and cell death. Gemcitabine is a prodrug so activity occurs as a result of intracellular conversion to two active metabolites, gemcitabine diphosphate and gemcitabine triphosphate by deoxycitidine kinase. Gemcitabine diphosphate also inhibits ribonucleotide reductase, the enzyme responsible for catalyzing synthesis of deoxynucleoside triphosphates required for DNA synthesis. Finally, Gemcitabine triphosphate (diflurorodeoxycytidine triphosphate) competes with endogenous deoxynucleoside triphosphates for incorporation into DNA.
Each capsule contains Hydroxyurea 500mg
L01XX05 – Hydroxycarbamide
An antineoplastic agent that inhibits DNA synthesis through the inhibition of ribonucleoside diphosphate reductase.
Hydroxyurea has dose-dependent synergistic activity with cisplatin in vitro. In vivo Hydroxyurea showed activity in combination with cisplatin against the LX-1 and CALU-6 human lung xenografts, but minimal activity was seen with the NCI-H460 or NCI-H520 xenografts. Hydroxyurea was synergistic with cisplatin in the Lewis lung murine xenograft. Sequential exposure to Hydroxyurea 4 hours before cisplatin produced the greatest interaction.
Hydroxyurea is converted to a free radical nitroxide (NO) in vivo, and transported by diffusion into cells where it quenches the tyrosyl free radical at the active site of the M2 protein sub unit of ribonucleotide reductase, inactivating the enzyme. The entire replicase complex, including ribonucleotide reductase, is inactivated and DNA synthesis is selectively inhibited, producing cell death in S phase and synchronization of the fraction of cells that survive. Repair of DNA damaged by chemicals or irradiation is also inhibited by hydroxyurea, offering potential synergy between hydroxyurea and radiation or alkylating agents. Hydroxyurea also increases the level of fetal hemoglobin, leading to a reduction in the incidence of vasoocclusive crises in sickle cell anemia. Levels of fetal hemoglobin increase in response to activation of soluble guanylyl cyclase (sGC) by hydroxyurea-derived NO.
Each vial contains zoledronic acid (as zoledronic acid monohydrate) – 4 mg.
M05BA08 — Zoledronic acid
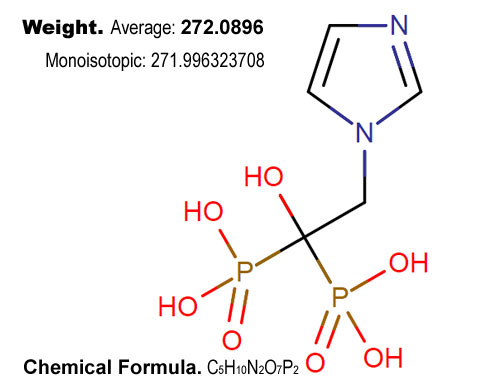
Zoledronic acid is a bisphosphonate and used to prevent skeletal fractures in patients with cancers such as multiple myeloma and prostate cancer. It can also be used to treat hypercalcemia of malignancy and can be helpful for treating pain from bone metastases.
An annual dose of Zoledronate may also prevent recurring fractures in patients with a previous hip fracture.
Zoledronate is a bisphosphonic acid which is an inhibitor of osteoclastic bone resorption. Zoledronate is used to prevent osteoporosis and skeletal fractures, particularly in patients with cancers such as multiple myeloma and prostate cancer. It can also be used to treat hypercalcemia, particularly hypercalcemia of malignancy. It can also be helpful for treating pain from bone metastases.
The action of zoledronate on bone tissue is based partly on its affinity for hydroxyapatite, which is part of the mineral matrix of bone. Zoledronate also targets farnesyl pyrophosphate (FPP) synthase. Nitrogen-containing bisphosphonates such as zoledronate appear to act as analogues of isoprenoid diphosphate lipids, thereby inhibiting FPP synthase, an enzyme in the mevalonate pathway. Inhibition of this enzyme in osteoclasts prevents the biosynthesis of isoprenoid lipids (FPP and GGPP) that are essential for the post-translational farnesylation and geranylgeranylation of small GTPase signalling proteins. This activity inhibits osteoclast activity and reduces bone resorption and turnover. In postmenopausal women, it reduces the elevated rate of bone turnover, leading to, on average, a net gain in bone mass.

L01BA01 — Methotrexate
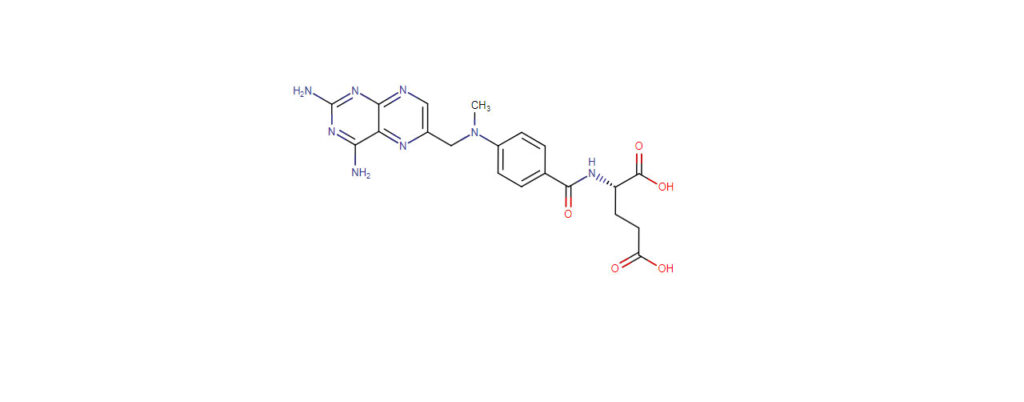
Methotrexate is a folate derivative that inhibits several enzymes responsible for nucleotide synthesis. This inhibition leads to suppression of inflammation as well as prevention of cell division. Because of these effects, methotrexate is often used to treat inflammation caused by arthritis or to control cell division in neoplastic diseases such as breast cancer and non-Hodgkin’s lymphoma.
Due to the toxic effects of methotrexate, it is indicated for treatment of some forms of arthritis and severe psoriasis only if first line treatment has failed or patients are intolerant of those treatments.
Methotrexate inhibits enzymes responsible for nucleotide synthesis which prevents cell division and leads to anti-inflammatory actions. It has a long duration of action and is generally given to patients once weekly. Methotrexate has a narrow therapeutic index.
L01AA01 — Cyclophosphamide
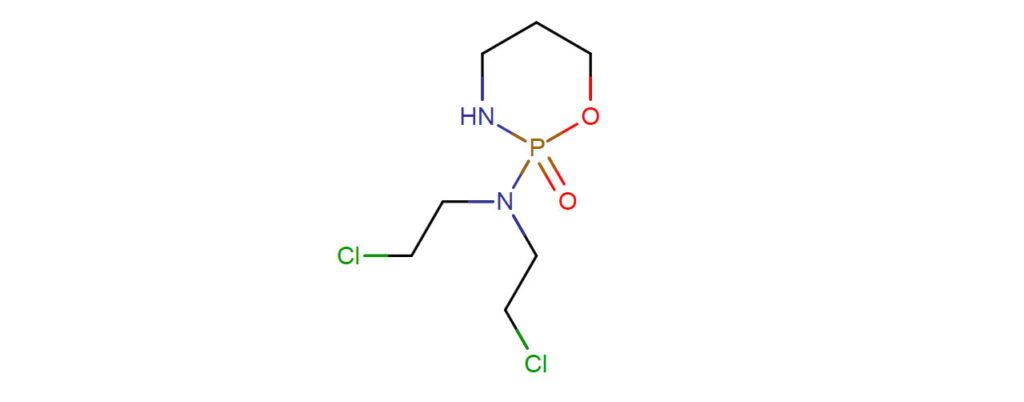
Precursor of an alkylating nitrogen mustard antineoplastic and immunosuppressive agent that must be activated in the liver to form the active aldophosphamide. It has been used in the treatment of lymphoma and leukemia. Its side effect, alopecia, has been used for defleecing sheep. Cyclophosphamide may also cause sterility, birth defects, mutations, and cancer.
Cyclophosphamide is an antineoplastic in the class of alkylating agents and is used to treat various forms of cancer. Alkylating agents are so named because of their ability to add alkyl groups to many electronegative groups under conditions present in cells. They stop tumor growth by cross-linking guanine bases in DNA double-helix strands – directly attacking DNA. This makes the strands unable to uncoil and separate. As this is necessary in DNA replication, the cells can no longer divide. In addition, these drugs add methyl or other alkyl groups onto molecules where they do not belong which in turn inhibits their correct utilization by base pairing and causes a miscoding of DNA. Alkylating agents are cell cycle-nonspecific. Alkylating agents work by three different mechanisms all of which achieve the same end result – disruption of DNA function and cell death.
Alkylating agents work by three different mechanisms:
1) attachment of alkyl groups to DNA bases, resulting in the DNA being fragmented by repair enzymes in their attempts to replace the alkylated bases, preventing DNA synthesis and RNA transcription from the affected DNA,
2) DNA damage via the formation of cross-links (bonds between atoms in the DNA) which prevents DNA from being separated for synthesis or transcription, and 3) the induction of mispairing of the nucleotides leading to mutations.
Each vial contains oxaliplatin (50 mg or 100 mg)
L01XA03 — Oxaliplatin
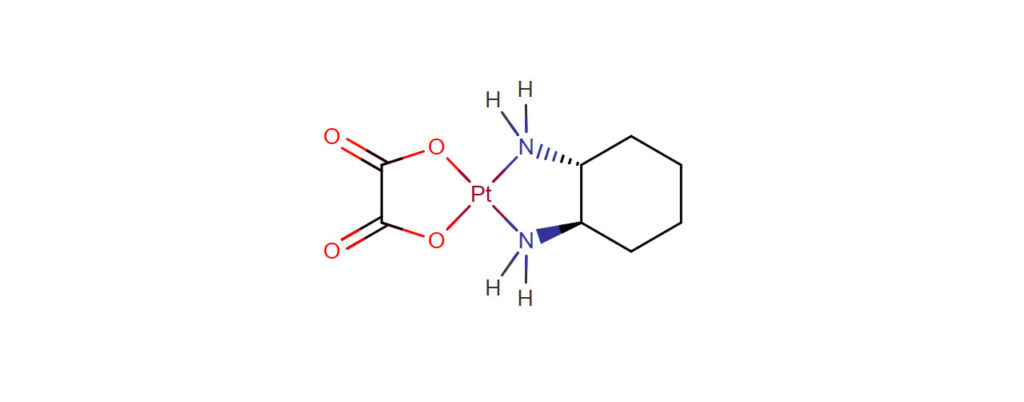
Oxaliplatin is a platinum-based chemotherapy drug in the same family as cisplatin and carboplatin. It is typically administered in combination with fluorouracil and leucovorin in a combination known as Folfox for the treatment of colorectal cancer. Compared to cisplatin the two amine groups are replaced by cyclohexyldiamine for improved antitumour activity. The chlorine ligands are replaced by the oxalato bidentate derived from oxalic acid in order to improve water solubility.
Oxaliplatin selectively inhibits the synthesis of deoxyribonucleic acid (DNA). The guanine and cytosine content correlates with the degree of Oxaliplatin-induced cross-linking. At high concentrations of the drug, cellular RNA and protein synthesis are also suppressed.
Oxaliplatin undergoes nonenzymatic conversion to active derivatives via displacement of the labile oxalate ligand. Several transient reactive species are formed, including monoaquo and diaquo DACH platinum, which covalently bind with macromolecules. After activation, oxaliplatin binds preferentially to the guanine and cytosine moieties of DNA, leading to cross-linking of DNA, thus inhibiting DNA synthesis and transcription. Cytotoxicity is cell-cycle nonspecific.

each capsule contains aprepitant – 125mg and 80mg.
A04AD12 — Aprepitant
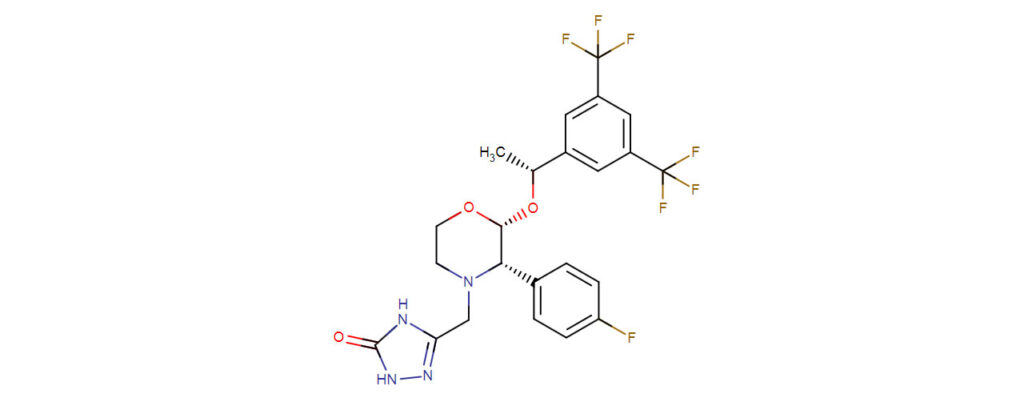
Aprepitant, an antiemetic, is a substance P/neurokinin 1 (NK1) receptor antagonist which, in combination with other antiemetic agents, is indicated for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy. Aprepitant is a selective high-affinity antagonist of human substance P/neurokinin 1 (NK1) receptors. Aprepitant has little or no affinity for serotonin (5-HT3), dopamine, and corticosteroid receptors, the targets of existing therapies for chemotherapy-induced nausea and vomiting (CI NV).
Aprepitant, an antiemetic, is a substance P/neurokinin 1 (NK1) receptor antagonist which, in combination with other antiemetic agents, is indicated for the prevention of acute and delayed nausea and vomiting associated with initial and repeat courses of highly emetogenic cancer chemotherapy. Aprepitant is a selective high-affinity antagonist of human substance P/neurokinin 1 (NK1) receptors. Aprepitant has little or no affinity for serotonin (5-HT3), dopamine, and corticosteroid receptors, the targets of existing therapies for chemotherapy-induced nausea and vomiting (CI NV).
Aprepitant has been shown in animal models to inhibit emesis induced by cytotoxic chemotherapeutic agents, such as cisplatin, via central actions. Animal and human Positron Emission Tomography (PET) studies with Aprepitant have shown that it crosses the blood brain barrier and occupies brain NK1 receptors. Animal and human studies show that Aprepitant augments the antiemetic activity of the 5-HT3-receptor antagonist ondansetron and the corticosteroid ethasone and inhibits both the acute and delayed phases of cisplatin induced emesis.
Each film coated tablet contains Deferiprone 500mg
V03AC02 — Deferiprone
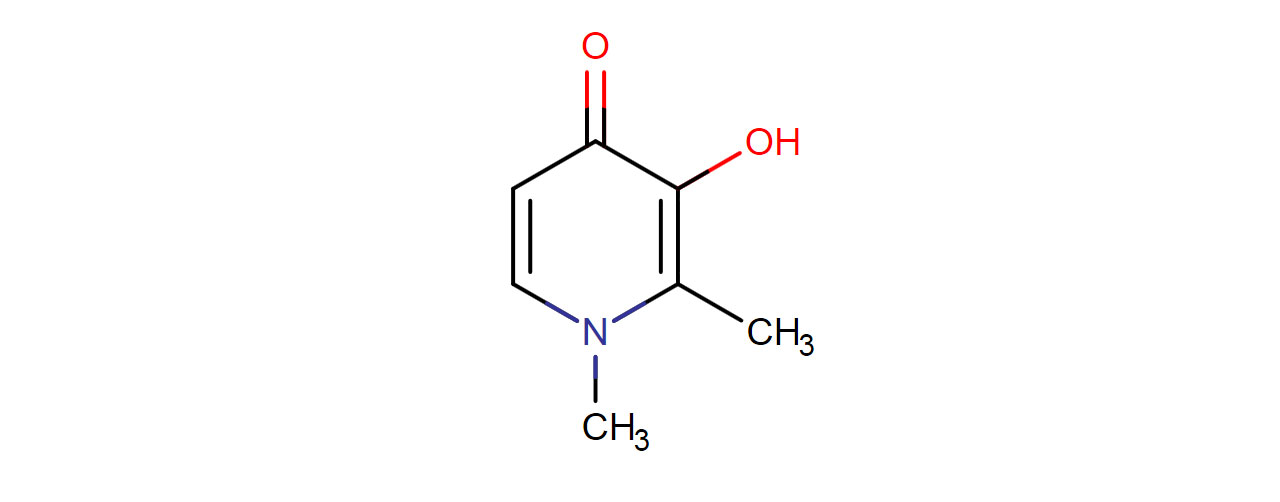
Deferiprone is an oral iron chelator used as a second line agent in thalassemia syndromes when iron overload from blood transfusions occurs. Thalassemias are a type of hereditary anaemia due a defect in the production of hemoglobin. As a result, erythropoiesis, the production of new red blood cells, is impaired.
Deferiprone is an iron chelator that binds to ferric ions (iron III) and forms a 3:1 (deferiprone:iron) stable complex and is then eliminated in the urine. Deferiprone is more selective for iron in which other metals such as zinc, copper, and aluminum have a lower affinity for deferiprone.

Each vial contains Decarbazine 200 mg
L01AX04 — Dacarbazine
Bicalutamide is an oral non-steroidal anti-androgen for prostate cancer. It is comprised of a racemic mixture that is a 50:50 composition of the (R)-bicalutamide and (S)-bicalutamide enantionmers. Bicalutamide binds to the androgen receptor.
Bicalutamide is an antineoplastic hormonal agent primarily used in the treatment of prostate cancer. Bicalutamide is a pure, nonsteroidal anti-androgen with affinity for androgen receptors (but not for progestogen, estrogen, or glucocorticoid receptors). Consequently, Bicalutamide blocks the action of androgens of adrenal and testicular origin which stimulate the growth of normal and malignant prostatic tissue. Prostate cancer is mostly androgen-dependent and can be treated with surgical or chemical castration. To date, antiandrogen monotherapy has not consistently been shown to be equivalent to castration.
Bicalutamide competes with androgen for the binding of androgen receptors, consequently blocking the action of androgens of adrenal and testicular origin which stimulate the growth of normal and malignant prostatic tissue.